GATK pipeline
What is GATK?
The Genome Analysis Toolkit (GATK) is a software package developed by the Broad Institute specifically for the analysis of high-throughput sequencing data. It is designed to facilitate the discovery and genotyping of variants in genomic sequences, such as single nucleotide polymorphisms (SNPs) and insertions/deletions (indels).
Key Features of GATK
-
Robust Data Processing: The toolkit incorporates a series of best practices for data preprocessing, including alignment, duplicate marking, and quality score recalibration, ensuring high-quality input for variant calling.
-
Variant Discovery: GATK provides tools to identify genetic variants from raw sequencing data, enabling researchers to uncover differences in genomes.
-
Genotyping: After variants are called, GATK can also genotype these variants across multiple samples, providing insights into population genetics and disease association studies.
-
Flexible Workflow: GATK supports a wide range of workflows, from small-scale studies to large population genomics projects, and is compatible with various input formats, including BAM and VCF.
-
Comprehensive Documentation: GATK is accompanied by extensive documentation and tutorials, making it accessible to both novice and experienced researchers.
Why Use GATK?
- High Accuracy: GATK employs advanced statistical methods that enhance the accuracy of variant calling and reduce false positives.
- Community Support: As one of the most widely used tools in genomics, GATK has a large user community and frequent updates based on user feedback and advancements in the field.
- Integration with Other Tools: GATK can be easily integrated with other bioinformatics tools for downstream analysis, such as annotation and visualization.
Applications
GATK is used in various fields, including:
- Medical Genomics: Identifying genetic variants associated with diseases.
- Population Genetics: Studying genetic diversity and evolutionary patterns.
- Cancer Genomics: Detecting somatic mutations in tumor samples.
Theoretical Foundations of the GATK Pipeline
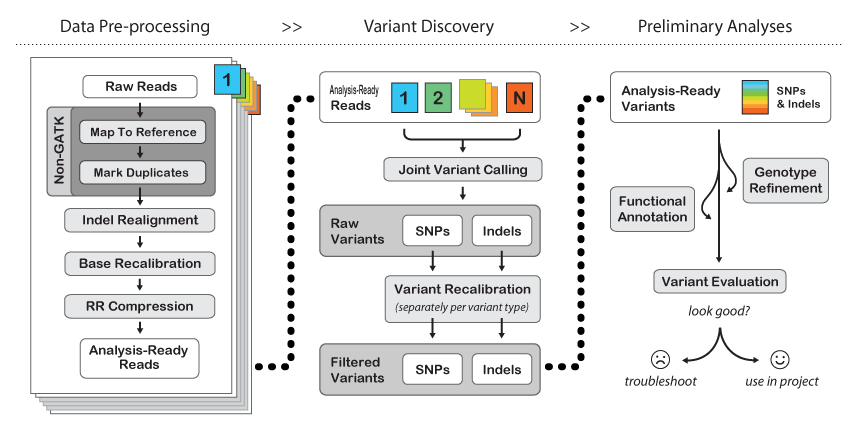
1. Data Preprocessing
Alignment
Alignment is the process of mapping sequencing reads to a reference genome. Accurate alignment is crucial, as it affects all subsequent analyses. Tools like BWA (Burrows-Wheeler Aligner) or Bowtie2 are commonly used for this step. The goal is to identify where each read originates from in the reference genome, accounting for potential sequencing errors and biological variations.
Marking Duplicates
During PCR amplification, some DNA fragments may be copied multiple times, resulting in duplicate reads. Marking these duplicates is essential to avoid overestimating variant frequencies. GATK's MarkDuplicates tool identifies and flags these duplicates based on their alignment coordinates.
Base Quality Score Recalibration (BQSR)
Quality scores provided by sequencing platforms may not accurately reflect the true error rates. BQSR adjusts these scores using machine learning models that consider covariates such as the context of the base and the read position. This recalibration enhances the reliability of subsequent variant calling.
2. Variant Calling
HaplotypeCaller
The HaplotypeCaller uses a haplotype-based approach to variant calling. Instead of calling variants for each individual read, it considers the entire local context of the reads. This method increases sensitivity and specificity, particularly in regions of the genome with complex variations. The output is typically a GVCF (Genome Variant Call Format) file, which provides information on both variants and non-variant regions.
GenotypeGVCFs
This step combines multiple GVCFs from different samples into a single VCF file. By using joint genotyping, the tool can leverage information from all samples, improving the accuracy of variant calls and allowing for better detection of rare variants.
3. Variant Filtering
Filtering is critical to eliminate false positive variant calls. GATK provides two main approaches:
- Hard Filters: Set specific thresholds for various quality metrics, such as depth of coverage, quality scores, and more.
- Variant Quality Score Recalibration (VQSR): A more sophisticated approach that uses machine learning to model the quality of variants based on known polymorphisms and various metrics.
4. Annotation
Annotation adds biological context to the variants. Tools like ANNOVAR and VEP can be integrated into the pipeline to provide information on the functional impact of variants, such as whether they affect coding sequences, regulatory regions, or are associated with known diseases.
5. Visualization
Visualization tools like IGV (Integrative Genomics Viewer) allow researchers to manually inspect variant calls in the context of the aligned reads. This step is crucial for validating variant calls and understanding the biological implications of identified variants.
Example Workflow
Step 1: Align reads
bwa mem reference.fasta reads.fastq > aligned.sam
Step 2: Convert SAM to BAM, sort, and mark duplicates
samtools view -Sb aligned.sam | samtools sort -o sorted.bam gatk MarkDuplicates -I sorted.bam -O dedup.bam -M metrics.txt
Step 3: Base Quality Score Recalibration
gatk BaseRecalibrator -I dedup.bam -R reference.fasta --known-sites known_sites.vcf -O recal_data.table gatk ApplyBQSR -I dedup.bam -R reference.fasta --bqsr-recal-file recal_data.table -O recal.bam
Step 4: Variant Calling
gatk HaplotypeCaller -R reference.fasta -I recal.bam -O raw_variants.g.vcf -ERC GVCF
Step 5: Genotype GVCFs
gatk GenotypeGVCFs -R reference.fasta -V raw_variants.g.vcf -O final_variants.vcf
Step 6: Filter variants
gatk VariantFiltration -V final_variants.vcf -O filtered_variants.vcf --filter-expression "QUAL < 30.0" --filter-name "LowQual"
Resources
- Ressource 1: GATK official documentation.